
ChExMix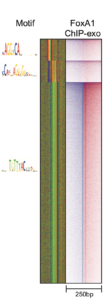

ChExMix aims to characterize protein-DNA binding subtypes in ChIP-exo experiments. ChExMix assumes that different regulatory complexes will result in different protein-DNA crosslinking signatures in ChIP-exo data, and thus analysis of ChIP-exo sequencing tag patterns should enable detection of multiple protein-DNA binding modes for a given regulatory protein. ChExMix uses a mixture modeling framework to probabilistically model the genomic locations and subtype membership of protein-DNA binding events, leveraging both ChIP-exo tag enrichment patterns and DNA sequence information. In doing so, ChExMix offers a more principled and robust approach to characterizing binding subtypes than simply clustering binding events using motif information.
 multiMDS
multiMDS
MultiMDS extends on the concepts in miniMDS to enable locus-specific structural comparisons of two Hi-C datasets. It jointly infers and aligns 3D structures from two datasets, such as different cell types. The output is aligned 3D structure files (which can be plotted, see below), locus-specific quantifications of relocalization, and compartment changes as a fraction of total relocalization. The amount of relocalization at each locus represents how much the locus changes between the datasets, which may be correlated with functional changes.
miniMDS
minMDS is a tool for inferring and plotting 3D structures using partitioned MDS, a novel approximation to multidimensional scaling (MDS). It produces a single 3D structure from a Hi-C BED file, representing an ensemble average of chromosome conformations within the population of cells. miniMDS is able to process high-resolution Hi-C data quickly with limited memory requirements. Human genome 3D structures can be inferred at kilobase-resolution within several hours on a desktop computer. miniMDS also supports inter-chromosomal structural inference.
SeqUnwinder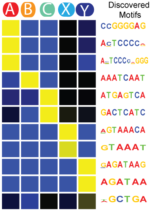
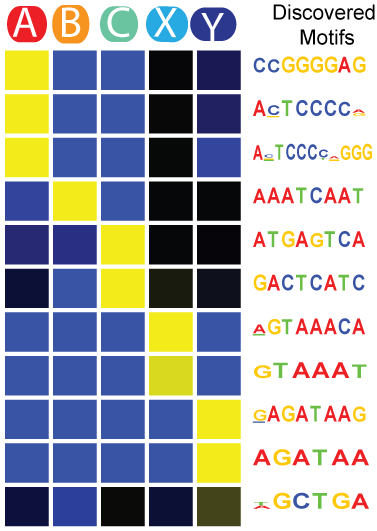
SeqUnwinder is a framework for characterizing class-discriminative motifs in a collection of genomic loci that have several (overlapping) annotation labels. SeqUnwinder treats motif-finding as a multi-label classification problem. Given a collection of genomic sequences, each labeled with one or more annotation labels, SeqUnwinder uses a multi-class logistic regression model to find motifs associated with each label. SeqUnwinder is useful in settings where overlapping sets of regulatory sites have been collected from multiple cell types, and we are interested in finding motifs that correspond to each cell type.
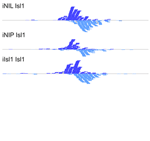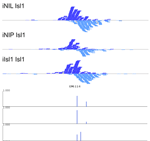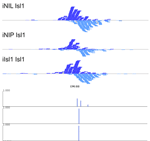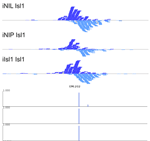 MultiGPS
MultiGPS
MultiGPS is a novel framework for analyzing collections of multi-condition ChIP-seq datasets and characterizing differential binding events between conditions. In analyzing multiple-condition ChIP-seq datasets, MultiGPS encourages consistency in the reported binding event locations across conditions and provides accurate estimation of ChIP enrichment levels at each event. MultiGPS manual and downloads are available here.
GPS and GEM
GPS & GEM predict protein-DNA interaction events at high spatial resolution from ChIP-seq or ChIP-exo data while retaining the ability to resolve closely spaced events that appear as a single cluster of reads. Both GPS and GEM model observed reads using a complexity penalized mixture model and efficiently predict event locations with a segmented EM algorithm. GPS was our first generation software package in this series. GEM extends the concept by linking binding event discovery and motif discovery with an integrated model of ChIP reads and proximal DNA sequences. GEM uses predicted binding events as a positional prior for a novel k-mer based de novo motif discovery algorithm, and reciprocally improves the resolution of the binding event predictions using the discovered motifs as a positional prior.
STAMP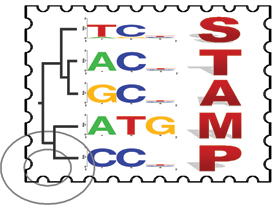
STAMP is a webserver resource for aligning transcription factor DNA-binding motifs. Input motifs may be aligned against each other using a wide choice of comparison metrics and alignment strategies. A multiple alignment, familial binding profile, and similarity tree are also produced from the set of input motifs. STAMP also matches each of the input motifs against a choice of databases of known TF binding motifs. STAMP allows the input of many different motif formats, including the input of entire output files from a number of supported motif-finders. In this way, STAMP provides a valuable resource for those researchers who wish to interpret their motif-finding results; such users may simply analyze their results using STAMP to see if any of their newly discovered motifs are similar to any known binding preferences.
Code for a command-line version of STAMP is available here: https://github.com/seqcode/stamp
SOMBRERO
SOMBRERO is a motif-finder that is based on the Self-Organizing Map neural network algorithm. In contrast to other probabilistic motif discovery tools, SOMBRERO poses motif-finding as a clustering problem. As such, SOMBRERO simultaneously estimates all motif signals in the input sequences (regulatory signals are separated from others during post-processing), as opposed to estimating each significant signal one-by-one. This clustering approach to motif-finding is undoubtedly more computationally costly than more traditional approaches. However, the great advantage of the approach is that multiple instances of prior knowledge may be used to initialize the motif-search. Prior knowledge of the implanted motif has been shown to significantly improve the accuracy of motif-finders. Of course, in typical de novo motif searches, we do not know what type of signal we are looking for. Traditional motif-finders may only incorporate one prior at a time, so the application of priors to motif-finding has been limited to those rare cases where certain motif signals are expected. SOMBRERO is the first motif-finder that can incorporate knowledge of all known motifs at the start of the motif search.
RescueNet
RescueNet uses the Self-Organizing Map neural network algorithm for codon usage anaysis and gene-prediction. In its gene prediction functionality, RescueNet can estimate multiple models of gene codon usage properties during training. This offers advantageous gene-finding performance in cases where a diverse number of codon usage patterns are displayed. Examples include metagenomic datasets and prokaryotic genomes where mutational pressure, translational efficiency and horizontal gene transfer have diversified the displayed codon usage patterns.